全部商品分类
- 手术急救设备
- 无影灯 急救车 电动手术台 电动吸引器 担架 手术套车 吸痰器 麻醉机 电动呼吸机 婴儿培养箱 注射泵 电动牵引床 输液泵 电动洗胃机 负压调整器 脚踏吸引器 手术导航系统 化疗泵 排痰机 心肺复苏器


[推荐理由] 斯曼峰LX-3妇科吸引器是一款电动人流吸引器,供医务人员对早期妊赈的孕妇进行人流吸引手术用,是妇科手术的理想设备。

[推荐理由]GD350-B5型多功能高频电刀拥有九种功能模式,两路手控、两路脚控共四路输出,适用于各科室的大小手术,还能结合氩气工作站组成先进的氩气高频电刀系统,提升手术效果和改善手术环境,是沪通高频电刀中的旗舰机型。
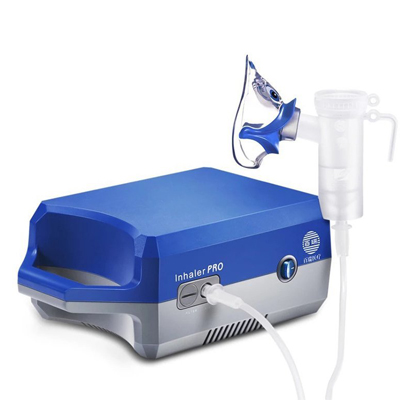
国产百瑞压缩式雾化器操作简便,可自行安装使用,雾化器配有口含器、弯接管、成人及儿童面罩,适合不同人群吸入治疗,百瑞雾化器根据患者吸气量的变化,自动调节药物的输出率。

>上海蓝德双通道注射泵LD-P2020II可以自动检测注射泵型号、输液总量显示、流速显示、电源指示、残余量可调、压力等级可调、流速等级可调、报警消音、报警音量可调等功能。

YX980D型电动吸引器是一种高负压、大流量的医用吸引器。可在数秒内建立起所需要的负压,适合需要快速吸出大量渗出液的场合。安全可靠的控制系统,是各医疗单位的理想选择。

DXW-D型洗胃机除了蠕动泵等固有特点外,还能对进液量进行选择,并可实现自动、手动两种洗胃模式及压力、液量双重保护、洗胃状态显示等功能,有效提高洗胃工作效率。


































 400-006-1850
400-006-1850 沪公网安备 31011502008791号
沪公网安备 31011502008791号